
Computational Catalysis
Computational Catalysis
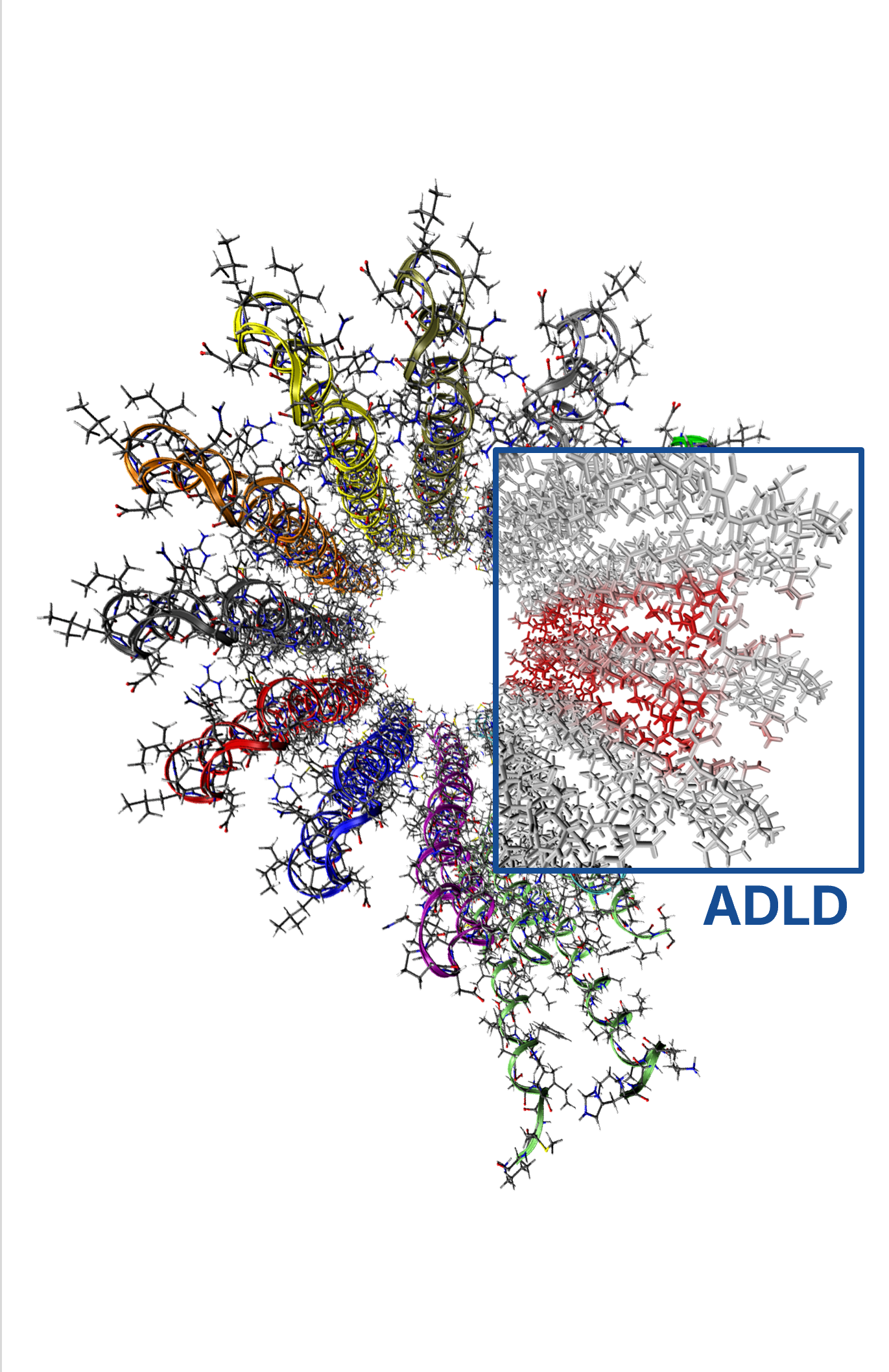
Catalysis is a cornerstone of modern chemical research, playing a role in the production of over 80% of all manufactured goods. Yet, despite advances in theory and computation, the number of catalyst predictions that successfully translate into experimental discovery remains limited. In our group, we aim to bridge this gap by combining state-of-the-art computational tools to elucidate the mechanisms of complex catalytic reactions and the electronic structure of key intermediates.
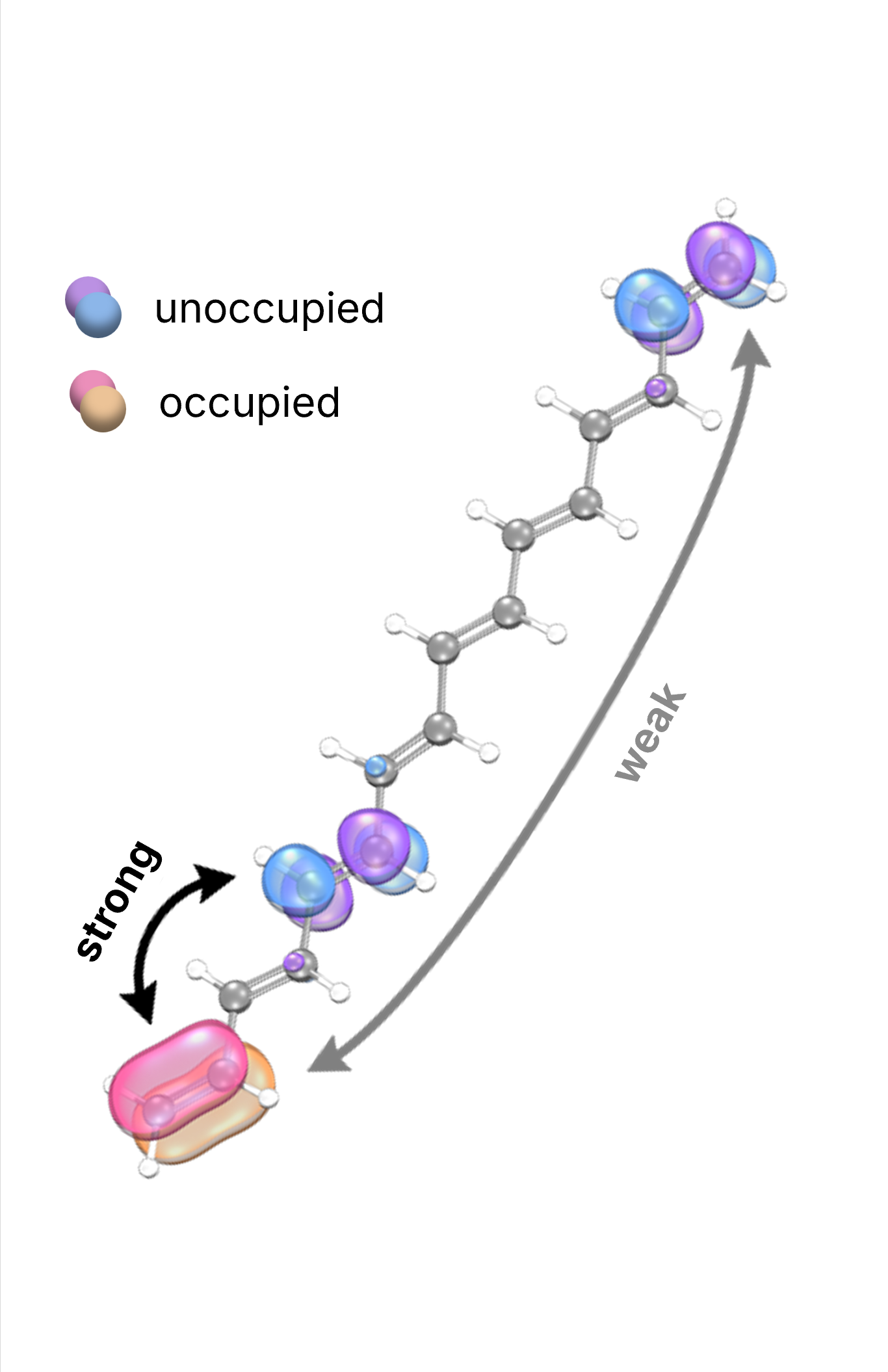
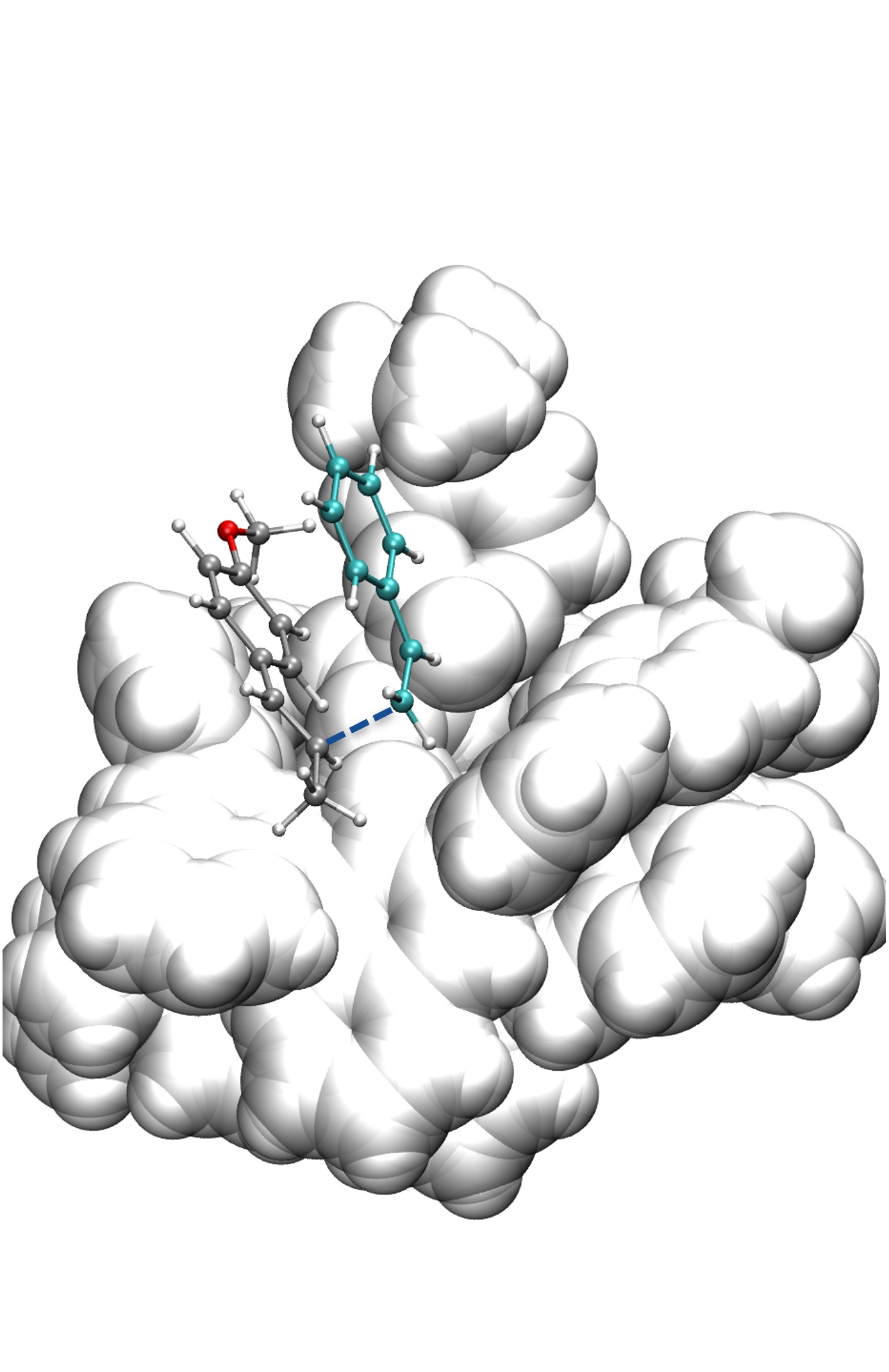
Our approach integrates advanced methodologies, including novel local coupled cluster techniques, enhanced conformational sampling algorithms, modern exchange–correlation functionals, machine learning techniques, energy decomposition and bond analysis schemes, quantum embedding frameworks, and both implicit and explicit solvation models. These tools allow us to deliver detailed, predictive insights into catalysis at the atomic level.
Our work targets the development of new synthetic processes with industrial relevance, with applications in:
(i) asymmetric organo- and transition metal catalysis,
(ii) electrocatalytic hydrogen and oxygen evolution reactions, and
(iii) biocatalysis.
This research is conducted in close collaboration with experimental groups, enabling a feedback loop between computation and synthesis.